发布时间:2026-05-24
近日,中国科学院广州生物医药与健康研究院(以下简称“广州健康院”) 细胞数字孪生研究组在《Briefings in Bioinformatics》上发表了题为“scMarkerGene: an interpretable neural network framework for cell-type-specific marker gene discovery”的研究论文。
细胞类型的精准识别是单细胞转录组学分析的基础,而细胞类型特异性标记基因(marker gene)的发现则是这一过程的核心。传统方法多依赖统计阈值或聚类启发式策略,易受数据噪声、注释偏差及基因高表达但非特异性等问题的干扰。针对上述挑战,研究团队提出了scMarkerGene——一个基于可解释神经网络框架的细胞类型特异性标记基因发现模型。scMarkerGene通过构建“贡献分数矩阵”,将神经网络模型中每个基因对细胞类型判别的影响量化为可解释的贡献值,并结合集成学习与特异性过滤策略,实现了对不同物种、不同测序技术、不同细胞群体规模及高稀疏性数据的稳健标记基因识别。
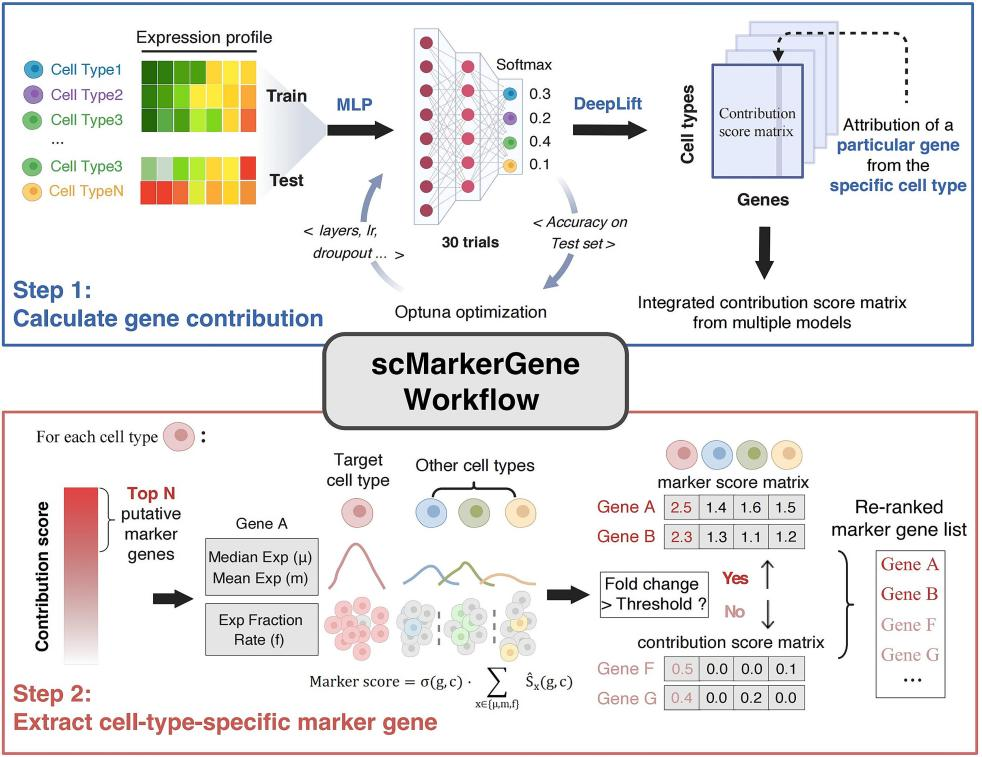
scMarkerGene的工作流程主要包括两个步骤(如图1):第一步:贡献分数计算。基于多层感知机(MLP)构建分类模型,通过集成多个超参数扰动训练得到的模型,利用DeepLIFT解释方法计算每个基因对细胞类型判别的贡献分数,并经过统计检验筛选出候选标记基因。第二步:特异性筛选与重排序。在候选基因基础上,结合基因的均值表达、中位数表达及检出率,构建“marker评分”,并与轮廓系数加权后对基因进行重排序,最终输出高特异性的细胞类型标记基因。
研究团队在10个公开的单细胞RNA测序数据集上对scMarkerGene进行了系统评估,涵盖拟南芥、果蝇、小鼠、人类等多个物种及多种测序平台。结果表明,scMarkerGene在log2FC、标准化Z-score等指标上均一致优于scanpy、scMAGs、SMaSH、scVI等现有方法。在模拟数据实验中,scMarkerGene识别高特异性标记基因,显著领先其他方法,并能有效滤除非特异性基因。在引入不同比例的随机丢失噪声后,scMarkerGene依然保持高鲁棒性,而同类方法SMaSH的性能则明显下降。在骨类器官数据集中,scMarkerGene在粗粒度与细粒度细胞类型上均能稳定识别高特异性标记基因,尤其在样本量不足100个细胞的小群体中仍保持最高log2FC,展现出对罕见细胞群体的强大适应性。
此外,在空间转录组与拟时间序列分析中,scMarkerGene识别出的标记基因在10X Visium小鼠脑组织及人黑色素瘤数据中均展现出清晰的空间定位特征;在BEELINE基准数据中,其在不同离散时间状态下预测的标记基因也均取得最高的log2FC值。
scMarkerGene区别传统方法依赖表达均值差异检验方式,以判别函数为核心,以贡献分数为统一度量,建立基因贡献分数与分类决策边界敏感度之间的数学联系,推动标记基因筛选从“统计描述”走向“机制解析”,为从复杂单细胞数据中解析细胞身份提供了可靠方法。
广州健康院赵永兵研究员为该论文的唯一通讯作者。博士后张景恺、高诗铠为该论文共同第一作者。该研究工作得到了中国国家重点研发计划以及广东省科技计划项目的资助支持。
图1. scMarkerGene工作流程
附件下载: